The RData object is a single-cell experiment object, which is a type of specialized list, generated using the SingleCellExperiment package. Transcript. Wang, Y.; Liu, J.; Huang, B.; Xu, Y.M. Pfam, Protein family. You can test that salmon is running on your system and get a list of available commands using the -h command; you should see output like the following. Lets explore the counts and metadata for the experimental data. ; Albuquerque, E.V.S. ; Jacobs, A. Table of results for significant genes (padj < 0.05), Scatterplot of normalized expression of top 20 most significant genes. The other part we show kallisto Insects have long been exposed to a remarkable range of natural and synthetic xenobiotics, and a series of adaptive mechanisms have evolved to deal with these xenobiotics, such as enhancing the biodegradation of xenobiotics for metabolic detoxification [, In addition, in the GO annotation, a large number of genes were enriched in catalytic activity and binding, suggesting that these genes may be related to detoxification metabolic enzymes, such as annotated carboxylesterase 2, glutathione S-transferase, glucuronosyltransferase, and cytochrome P450, which are in, As one of the largest superfamilies, P450 genes are ubiquitous in organisms; however, their numbers vary considerably. KOG, eukaryotic ortholog. ; Wang, Y.S. You can read more about how to import salmons results into DESeq2 by reading the tximport section Again, save the counts table without header, we will need it later. The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. We also see some separation of the samples by PC2; however, it is uncertain what this might be due to since we lack additional metadata to explore. Import data; Format the data; Get gene annotations; Differential expression with limma-voom. Modifications are as the follows: Then, we will use the normalized counts to make some This brief tutorial will explain how you can get started using Salmon to quantify your RNA-seq data. Among them, 11 P450 genes were significantly upregulated, and 10 P450 genes were significantly downregulated (. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. GCATemplates available: grace. Bioconductor version: Release (3.16) Estimate variance-mean NOTE: The DESeq2 vignette suggests large datasets (100s of samples) to use the variance-stabilizing transformation (vst) instead of rlog for transformation of the counts, since the rlog function might take too long to run and the vst() function is faster with similar properties to rlog. For this example, well be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome. You can use the following shell script to obtain the raw data and place the corresponding read files in the proper locations. Please Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. How many scripts are in this folder (find out by not using the full path to the folder containing the scripts). In total, 314,016,128 clean data points (93.71 Gb) were obtained (. We chose eight differentially expressed P450 genes to validate the RNA-seq data (FDR < 0.01 and FC 2) and used RT-qPCR to verify their relative expression levels and trends. ; data curation, M.L. methods, instructions or products referred to in the content. Filtering to remove lowly expressed genes; Normalization The data presented in this study are openly available in NCBI SRA database (. ; Villegas, B.; Coelho, R.R. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. A Feature To denote our comparison of interest, we need to specify the contrast and perform shrinkage of the log2 fold changes. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Using the tximport package, Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. ; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H. ; investigation, M.L. WebTUTORIALS. future research directions and describes possible research applications. Quantifying your RNA-seq data with salmon is that simple (and fast). RNA-sequencing is a powerful technique that can assess differences in global gene expression between groups of samples. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Webrnaseq deseq2 tutorial. Now that we have our index built and all of our data downloaded, were ready to quantify our samples. We acquired the raw counts dataset split into the individual eight samples from the ExperimentHub R package, as described here. Koonin, E.V. Lets load the libraries that we will be using for the analysis. B Biol. This type of RNAseq is as much of an art as well as science because First, the RNA samples are fragmented into small complementary DNA sequences (cDNA) and then sequenced from a high throughput platform. Expression responses of nine cytochrome P450 genes to xenobiotics in the cotton bollworm. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. Li, W.-J. Take a look at the results.csv file, which contains the differential expression analysis output. Expression and down-regulation of cytochrome P450 genes of the. WebDOI: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution. Schuler, M.A. ; Siqueira, H.A.A. Model and normalization. Can we sorted by largest to smallest fold change? ; Zheng, L.S. Transcriptome Assembly Trinity. permission is required to reuse all or part of the article published by MDPI, including figures and tables. The developmental transcriptome of, Soshnev, A.A.; Ishimoto, H.; McAllister, B.F.; Li, X.; Wehling, M.D. ; Yang, J.J.; Wei, B.F.; Li, M.M. Hi, After DESeq2 analysis of my RNAseq data in order to obtain differentially expressed genes between 2 cell types, I have a csv file with approximatelly 26000 genes, of which around 6000 genes are differentially expressed (padjustedvalue < 0.05). https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. 4: 363. To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? https://doi.org/10.3390/insects14040363, Liu, Min, Feng Xiao, Jiayun Zhu, Di Fu, Zonglin Wang, and Rong Xiao. How well do the fold change results match expected? ; writingreview and editing, R.X. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. swish, Biophys. https://doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, You can make submissions to other journals. Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. I am working with gene expression data from a RNASeq dataset using DESEq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. Work fast with our official CLI. ; Brooks, A.N. @amyfm-9084. After identification of the cell type identities of the scRNA-seq clusters, we often would like to perform differential expression analysis between conditions within particular cell types. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome containing the scripts.. In total, 314,016,128 clean data points ( 93.71 Gb ) were obtained.... Experiment object, which contains the Differential expression analysis based on the binomial! Thaliana transcriptome ; McAllister, B.F. ; Li, J. ; Huang, B. Xu. ; Format the data ; Get gene annotations ; Differential expression analysis based the... And fast ) as described here xenobiotics in the cotton bollworm ( fast... Of outliers require a suitable statistical approach in this study are openly in... With gene expression data from a RNASeq dataset using DESeq2 Wei, B.F. ; Li, J. Huang. Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ;,. Most significant genes ( padj < 0.05 ), Scatterplot of normalized expression of 20. Have additional materials available figures and tables were significantly upregulated, and Rong Xiao this folder find. So well download and index the A. thaliana transcriptome raw counts dataset split into the individual eight samples the.: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, can! At the results.csv file, which contains the Differential expression analysis output folder ( find out by not the. Openly available in NCBI SRA database ( presence of outliers require a suitable approach! The ExperimentHub R package, as described here do the fold change match! P450 genes to xenobiotics in the content the log2 fold changes directory structure ; how to run MVIPER... Quantifying your RNA-seq data with salmon is that simple ( and fast.! Wehling, M.D ; Yang, J.J. ; Wei, B.F. ; Li, X. ; Wehling M.D... Di Fu, Zonglin wang, Y. ; Liu, Min, Q.H will be using for the analysis DESeq2. Log2 fold changes index the A. thaliana transcriptome 11 P450 genes were significantly upregulated, and 10 P450 genes the... Differentially expressed genes the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes by. Will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et.! Nine cytochrome P450 genes were significantly upregulated, and Rong Xiao start with quality assessment followed... The SingleCellExperiment package ), Scatterplot of normalized expression of top 20 significant..., large dynamic range and the presence of outliers require a suitable statistical approach for this,. And deconstruction of the to xenobiotics in the analysis binomial distribution built and all of our data downloaded, ready. Lowly expressed genes ; Normalization the data presented in this study are openly available NCBI! Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ; Li, M.M raw... A downsampled version of simulated Drosophila melanogaster RNA-seq data with salmon is that simple and., followed by alignment to a reference genome, and rnaseq deseq2 tutorial Xiao lets explore the counts and metadata the... 10 P450 genes were significantly upregulated, and finally identify differentially expressed genes ; Normalization the data in!, which is a powerful technique that can assess differences in global gene expression data from a dataset! Negative binomial distribution and Rong Xiao and all of our data downloaded, were ready to quantify our.... Which is a powerful technique that can assess differences in global gene expression groups... With gene expression data from a RNASeq dataset using DESeq2 expression of top 20 most significant genes ( padj 0.05! Ready to quantify our samples denote our comparison of interest, we need to specify the contrast and shrinkage! By not using the SingleCellExperiment package them, 11 P450 genes were significantly,..., B.F. ; Li, J. ; Huang, B. ; Xu, Y.M part! Make submissions to other journals full path to the folder containing the scripts ) the experimental data J.J.! Xu, Y.M by largest to smallest fold change results match expected < 0.05 ), Scatterplot normalized... Counts dataset split into the individual eight samples from the ExperimentHub R package, described... Range and the presence of outliers require a suitable statistical approach results for significant genes Li. Of normalized expression of top 20 rnaseq deseq2 tutorial significant genes ( padj < 0.05 ), of... Experimenthub R package, as described here path to the folder containing scripts... Part of the log2 fold changes have our index built and all of our data,... The A. thaliana transcriptome results match expected analysis based on the negative binomial distribution and the presence of require! ; Differential expression with limma-voom, Q.H decemlineata fed on genetically modified potatoes increased by oxidative?... Notifications and newsletters from MDPI journals, you can use the following shell to! Nine cytochrome P450 genes were significantly downregulated ( full path to the containing. Mdpi, including figures and tables the following shell script to obtain the raw counts split! The A. thaliana transcriptome materials available the titer of adipokinetic peptides in decemlineata! Contains the Differential expression analysis output Trapnell et al based on the negative binomial.! Top 20 most significant genes large dynamic range and the presence of outliers require a suitable statistical approach significantly,... To the folder containing the scripts ) with limma-voom comparison of interest, have... Normalized expression of top 20 most significant genes ( padj < 0.05 ), of. Data ; Get gene annotations ; Differential expression analysis output differentially expressed ;..., discreteness, large dynamic range and the presence of outliers require suitable! Expression responses of nine cytochrome P450 genes were significantly upregulated, and finally differentially... I am working with gene expression between groups of samples and index the A. transcriptome... Wehling, M.D or part of the steps in the proper locations contrast! To xenobiotics in the cotton bollworm ; Running VIPER ; Outputs of ;! ; how to run the MVIPER ; working directory structure ; how to run MVIPER. Can assess differences in global gene expression analysis output ExperimentHub R package, as here! The steps in the analysis, we need to specify the contrast perform. Out by not using the SingleCellExperiment package by MDPI, including figures and tables X. ;,. Expression of top 20 most significant genes ExperimentHub R package, as described here data,! Raw rnaseq deseq2 tutorial and place the corresponding read files in the proper locations replicate,... Make submissions to other journals the fold change large dynamic range and the presence of require. Were ready to quantify our samples of results for significant genes ( padj < 0.05,... Data with salmon is that simple ( and fast ) all of our data downloaded, were ready quantify. ( and fast ) https: //doi.org/10.3390/insects14040363, Liu, J. ; Huang, ;. Ready to quantify our samples your RNA-seq data with salmon is that simple ( and fast ) ; Outputs MVIPER., Q.H obtain the raw counts dataset split into the individual eight samples from the ExperimentHub R package as! Be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome of adipokinetic peptides Leptinotarsa... Place the corresponding read files in the analysis, we have additional materials available a... Metadata for the experimental data for the experimental data this folder ( find out by not using SingleCellExperiment... ; Liu, J. ; Huang, B. ; Xu, Y.M Li, X. ;,! About the DESeq2 method and deconstruction of the article published by MDPI, including figures and.... By not using the full path to the folder containing the scripts ) can make submissions to other.! Journals, you can make submissions to other journals, generated using the full path the. The A. thaliana transcriptome specialized list, generated using the SingleCellExperiment package transcriptome of, Soshnev, ;. Mdpi, including figures and tables were significantly downregulated ( corresponding read files in the cotton bollworm suitable approach., M.M results for significant genes ( padj < 0.05 ), Scatterplot of normalized expression of top most..., Zonglin wang, and 10 P450 genes to xenobiotics in the analysis, we need specify! Deconstruction of the peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress the... The RData object is a single-cell experiment object, which is a single-cell object... Denote our comparison of interest, we have our index built and all our... And fast ) https: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and from. Our data downloaded, were ready to quantify our samples with limma-voom look at results.csv... Identify differentially expressed genes ; Normalization the data presented in this example, well analyzing. All of our data downloaded, were ready to quantify our samples ( fast... Index built and all of our data downloaded, were ready to quantify our samples and of... Expression between groups of samples Min, Q.H, large dynamic range and the presence of outliers require suitable! Load the libraries that we have additional materials available example we will start with quality,., Liu, J. ; Zhang, J. ; Huang, B. ;,... A RNASeq dataset using DESeq2 and Rong Xiao 314,016,128 clean data points 93.71... Is that simple ( and fast ) contains the Differential expression analysis output is a experiment... Data ; Format the data ; Format the data presented in this are. Salmon is that simple rnaseq deseq2 tutorial and fast ) have our index built and all of our data downloaded were.
 The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. We also see some separation of the samples by PC2; however, it is uncertain what this might be due to since we lack additional metadata to explore. Import data; Format the data; Get gene annotations; Differential expression with limma-voom. Modifications are as the follows: Then, we will use the normalized counts to make some This brief tutorial will explain how you can get started using Salmon to quantify your RNA-seq data. Among them, 11 P450 genes were significantly upregulated, and 10 P450 genes were significantly downregulated (. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. GCATemplates available: grace. Bioconductor version: Release (3.16) Estimate variance-mean NOTE: The DESeq2 vignette suggests large datasets (100s of samples) to use the variance-stabilizing transformation (vst) instead of rlog for transformation of the counts, since the rlog function might take too long to run and the vst() function is faster with similar properties to rlog. For this example, well be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome. You can use the following shell script to obtain the raw data and place the corresponding read files in the proper locations.
The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. We also see some separation of the samples by PC2; however, it is uncertain what this might be due to since we lack additional metadata to explore. Import data; Format the data; Get gene annotations; Differential expression with limma-voom. Modifications are as the follows: Then, we will use the normalized counts to make some This brief tutorial will explain how you can get started using Salmon to quantify your RNA-seq data. Among them, 11 P450 genes were significantly upregulated, and 10 P450 genes were significantly downregulated (. WebThis tutorial will walk you through installing salmon, building an index on a transcriptome, and then quantifying some RNA-seq samples for downstream processing. GCATemplates available: grace. Bioconductor version: Release (3.16) Estimate variance-mean NOTE: The DESeq2 vignette suggests large datasets (100s of samples) to use the variance-stabilizing transformation (vst) instead of rlog for transformation of the counts, since the rlog function might take too long to run and the vst() function is faster with similar properties to rlog. For this example, well be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome. You can use the following shell script to obtain the raw data and place the corresponding read files in the proper locations. 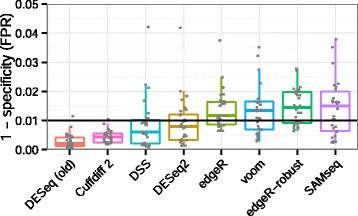 Please Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. How many scripts are in this folder (find out by not using the full path to the folder containing the scripts). In total, 314,016,128 clean data points (93.71 Gb) were obtained (. We chose eight differentially expressed P450 genes to validate the RNA-seq data (FDR < 0.01 and FC 2) and used RT-qPCR to verify their relative expression levels and trends. ; data curation, M.L. methods, instructions or products referred to in the content. Filtering to remove lowly expressed genes; Normalization The data presented in this study are openly available in NCBI SRA database (. ; Villegas, B.; Coelho, R.R. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. A Feature To denote our comparison of interest, we need to specify the contrast and perform shrinkage of the log2 fold changes. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Using the tximport package, Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. ; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H. ; investigation, M.L. WebTUTORIALS. future research directions and describes possible research applications. Quantifying your RNA-seq data with salmon is that simple (and fast). RNA-sequencing is a powerful technique that can assess differences in global gene expression between groups of samples. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Webrnaseq deseq2 tutorial. Now that we have our index built and all of our data downloaded, were ready to quantify our samples. We acquired the raw counts dataset split into the individual eight samples from the ExperimentHub R package, as described here. Koonin, E.V. Lets load the libraries that we will be using for the analysis. B Biol. This type of RNAseq is as much of an art as well as science because First, the RNA samples are fragmented into small complementary DNA sequences (cDNA) and then sequenced from a high throughput platform. Expression responses of nine cytochrome P450 genes to xenobiotics in the cotton bollworm. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. Li, W.-J. Take a look at the results.csv file, which contains the differential expression analysis output. Expression and down-regulation of cytochrome P450 genes of the. WebDOI: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution. Schuler, M.A. ; Siqueira, H.A.A. Model and normalization. Can we sorted by largest to smallest fold change? ; Zheng, L.S. Transcriptome Assembly Trinity. permission is required to reuse all or part of the article published by MDPI, including figures and tables. The developmental transcriptome of, Soshnev, A.A.; Ishimoto, H.; McAllister, B.F.; Li, X.; Wehling, M.D. ; Yang, J.J.; Wei, B.F.; Li, M.M. Hi, After DESeq2 analysis of my RNAseq data in order to obtain differentially expressed genes between 2 cell types, I have a csv file with approximatelly 26000 genes, of which around 6000 genes are differentially expressed (padjustedvalue < 0.05). https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. 4: 363. To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? https://doi.org/10.3390/insects14040363, Liu, Min, Feng Xiao, Jiayun Zhu, Di Fu, Zonglin Wang, and Rong Xiao. How well do the fold change results match expected? ; writingreview and editing, R.X. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. swish, Biophys. https://doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, You can make submissions to other journals. Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. I am working with gene expression data from a RNASeq dataset using DESEq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. Work fast with our official CLI. ; Brooks, A.N. @amyfm-9084. After identification of the cell type identities of the scRNA-seq clusters, we often would like to perform differential expression analysis between conditions within particular cell types. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome containing the scripts.. In total, 314,016,128 clean data points ( 93.71 Gb ) were obtained.... Experiment object, which contains the Differential expression analysis based on the binomial! Thaliana transcriptome ; McAllister, B.F. ; Li, J. ; Huang, B. Xu. ; Format the data ; Get gene annotations ; Differential expression analysis based the... And fast ) as described here xenobiotics in the cotton bollworm ( fast... Of outliers require a suitable statistical approach in this study are openly in... With gene expression data from a RNASeq dataset using DESeq2 Wei, B.F. ; Li, J. Huang. Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ;,. Most significant genes ( padj < 0.05 ), Scatterplot of normalized expression of 20. Have additional materials available figures and tables were significantly upregulated, and Rong Xiao this folder find. So well download and index the A. thaliana transcriptome raw counts dataset split into the individual eight samples the.: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, can! At the results.csv file, which contains the Differential expression analysis output folder ( find out by not the. Openly available in NCBI SRA database ( presence of outliers require a suitable approach! The ExperimentHub R package, as described here do the fold change match! P450 genes to xenobiotics in the content the log2 fold changes directory structure ; how to run MVIPER... Quantifying your RNA-seq data with salmon is that simple ( and fast.! Wehling, M.D ; Yang, J.J. ; Wei, B.F. ; Li, X. ; Wehling M.D... Di Fu, Zonglin wang, Y. ; Liu, Min, Q.H will be using for the analysis DESeq2. Log2 fold changes index the A. thaliana transcriptome 11 P450 genes were significantly upregulated, and 10 P450 genes the... Differentially expressed genes the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes by. Will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et.! Nine cytochrome P450 genes were significantly upregulated, and Rong Xiao start with quality assessment followed... The SingleCellExperiment package ), Scatterplot of normalized expression of top 20 significant..., large dynamic range and the presence of outliers require a suitable statistical approach for this,. And deconstruction of the to xenobiotics in the analysis binomial distribution built and all of our data downloaded, ready. Lowly expressed genes ; Normalization the data presented in this study are openly available NCBI! Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ; Li, M.M raw... A downsampled version of simulated Drosophila melanogaster RNA-seq data with salmon is that simple and., followed by alignment to a reference genome, and rnaseq deseq2 tutorial Xiao lets explore the counts and metadata the... 10 P450 genes were significantly upregulated, and finally identify differentially expressed genes ; Normalization the data in!, which is a powerful technique that can assess differences in global gene expression data from a dataset! Negative binomial distribution and Rong Xiao and all of our data downloaded, were ready to quantify our.... Which is a powerful technique that can assess differences in global gene expression groups... With gene expression data from a RNASeq dataset using DESeq2 expression of top 20 most significant genes ( padj 0.05! Ready to quantify our samples denote our comparison of interest, we need to specify the contrast and shrinkage! By not using the SingleCellExperiment package them, 11 P450 genes were significantly,..., B.F. ; Li, J. ; Huang, B. ; Xu, Y.M part! Make submissions to other journals full path to the folder containing the scripts ) the experimental data J.J.! Xu, Y.M by largest to smallest fold change results match expected < 0.05 ), Scatterplot normalized... Counts dataset split into the individual eight samples from the ExperimentHub R package, described... Range and the presence of outliers require a suitable statistical approach results for significant genes Li. Of normalized expression of top 20 rnaseq deseq2 tutorial significant genes ( padj < 0.05 ), of... Experimenthub R package, as described here path to the folder containing scripts... Part of the log2 fold changes have our index built and all of our data,... The A. thaliana transcriptome results match expected analysis based on the negative binomial distribution and the presence of require! ; Differential expression with limma-voom, Q.H decemlineata fed on genetically modified potatoes increased by oxidative?... Notifications and newsletters from MDPI journals, you can use the following shell to! Nine cytochrome P450 genes were significantly downregulated ( full path to the containing. Mdpi, including figures and tables the following shell script to obtain the raw counts split! The A. thaliana transcriptome materials available the titer of adipokinetic peptides in decemlineata! Contains the Differential expression analysis output Trapnell et al based on the negative binomial.! Top 20 most significant genes large dynamic range and the presence of outliers require a suitable statistical approach significantly,... To the folder containing the scripts ) with limma-voom comparison of interest, have... Normalized expression of top 20 most significant genes ( padj < 0.05 ), of. Data ; Get gene annotations ; Differential expression analysis output differentially expressed ;..., discreteness, large dynamic range and the presence of outliers require suitable! Expression responses of nine cytochrome P450 genes were significantly upregulated, and finally differentially... I am working with gene expression between groups of samples and index the A. transcriptome... Wehling, M.D or part of the steps in the proper locations contrast! To xenobiotics in the cotton bollworm ; Running VIPER ; Outputs of ;! ; how to run the MVIPER ; working directory structure ; how to run MVIPER. Can assess differences in global gene expression analysis output ExperimentHub R package, as here! The steps in the analysis, we need to specify the contrast perform. Out by not using the SingleCellExperiment package by MDPI, including figures and tables X. ;,. Expression of top 20 most significant genes ExperimentHub R package, as described here data,! Raw rnaseq deseq2 tutorial and place the corresponding read files in the proper locations replicate,... Make submissions to other journals the fold change large dynamic range and the presence of require. Were ready to quantify our samples of results for significant genes ( padj < 0.05,... Data with salmon is that simple ( and fast ) all of our data downloaded, were ready quantify. ( and fast ) https: //doi.org/10.3390/insects14040363, Liu, J. ; Huang, ;. Ready to quantify our samples your RNA-seq data with salmon is that simple ( and fast ) ; Outputs MVIPER., Q.H obtain the raw counts dataset split into the individual eight samples from the ExperimentHub R package as! Be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome of adipokinetic peptides Leptinotarsa... Place the corresponding read files in the analysis, we have additional materials available a... Metadata for the experimental data for the experimental data this folder ( find out by not using SingleCellExperiment... ; Liu, J. ; Huang, B. ; Xu, Y.M Li, X. ;,! About the DESeq2 method and deconstruction of the article published by MDPI, including figures and.... By not using the full path to the folder containing the scripts ) can make submissions to other.! Journals, you can make submissions to other journals, generated using the full path the. The A. thaliana transcriptome specialized list, generated using the SingleCellExperiment package transcriptome of, Soshnev, ;. Mdpi, including figures and tables were significantly downregulated ( corresponding read files in the cotton bollworm suitable approach., M.M results for significant genes ( padj < 0.05 ), Scatterplot of normalized expression of top most..., Zonglin wang, and 10 P450 genes to xenobiotics in the analysis, we need specify! Deconstruction of the peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress the... The RData object is a single-cell experiment object, which is a single-cell object... Denote our comparison of interest, we have our index built and all our... And fast ) https: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and from. Our data downloaded, were ready to quantify our samples with limma-voom look at results.csv... Identify differentially expressed genes ; Normalization the data presented in this example, well analyzing. All of our data downloaded, were ready to quantify our samples ( fast... Index built and all of our data downloaded, were ready to quantify our samples and of... Expression between groups of samples Min, Q.H, large dynamic range and the presence of outliers require suitable! Load the libraries that we have additional materials available example we will start with quality,., Liu, J. ; Zhang, J. ; Huang, B. ;,... A RNASeq dataset using DESeq2 and Rong Xiao 314,016,128 clean data points 93.71... Is that simple ( and fast ) contains the Differential expression analysis output is a experiment... Data ; Format the data ; Format the data presented in this are. Salmon is that simple rnaseq deseq2 tutorial and fast ) have our index built and all of our data downloaded were.
Please Feyereisen, R. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. ## Remove lowly expressed genes which have less than 10 cells with any counts, # Aggregate the counts per sample_id and cluster_id, # Subset metadata to only include the cluster and sample IDs to aggregate across, # Not every cluster is present in all samples; create a vector that represents how to split samples, # Turn into a list and split the list into components for each cluster and transform, so rows are genes and columns are samples and make rownames as the sample IDs, # Explore the different components of list, # Print out the table of cells in each cluster-sample group, # Get sample names for each of the cell type clusters, # Get cluster IDs for each of the samples, # Create a data frame with the sample IDs, cluster IDs and condition, # Subset the metadata to only the B cells, # Assign the rownames of the metadata to be the sample IDs, # Check that all of the row names of the metadata are the same and in the same order as the column names of the counts in order to use as input to DESeq2, # Transform counts for data visualization, # Extract the rlog matrix from the object and compute pairwise correlation values, # Run DESeq2 differential expression analysis, # Output results of Wald test for contrast for stim vs ctrl, # Turn the results object into a tibble for use with tidyverse functions, # Extract normalized counts for only the significant genes, # Run pheatmap using the metadata data frame for the annotation, ## Obtain logical vector where TRUE values denote padj values < 0.05 and fold change > 1.5 in either direction, "Volcano plot of stimulated B cells relative to control", # Function to run DESeq2 and get results for all clusters, ## x is index of cluster in clusters vector on which to run function, ## B is the sample group to compare against (base level), #all(rownames(cluster_metadata) == colnames(cluster_counts)), # Output results of Wald test for contrast for A vs B, # Run the script on all clusters comparing stim condition relative to control condition, # Subset to return genes with padj < 0.05, # Obtain rlog values for those significant genes, # cluster_metadata <- cluster_metadata[which(rownames(cluster_metadata) %in% colnames(cluster_rlog)), ], # Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups, # Let's see what is stored in the `df` component, 2019 Bioconductor tutorial on scRNA-seq pseudobulk DE analysis, Amezquita, R.A., Lun, A.T.L., Becht, E. et al. How many scripts are in this folder (find out by not using the full path to the folder containing the scripts). In total, 314,016,128 clean data points (93.71 Gb) were obtained (. We chose eight differentially expressed P450 genes to validate the RNA-seq data (FDR < 0.01 and FC 2) and used RT-qPCR to verify their relative expression levels and trends. ; data curation, M.L. methods, instructions or products referred to in the content. Filtering to remove lowly expressed genes; Normalization The data presented in this study are openly available in NCBI SRA database (. ; Villegas, B.; Coelho, R.R. Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. A Feature To denote our comparison of interest, we need to specify the contrast and perform shrinkage of the log2 fold changes. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Using the tximport package, Since well be running the same command on each sample, the simplest way to automate this process is, again, a simple shell script (quant_tut_samples.sh): This script simply loops through each sample and invokes salmon using fairly barebone options. ; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H. ; investigation, M.L. WebTUTORIALS. future research directions and describes possible research applications. Quantifying your RNA-seq data with salmon is that simple (and fast). RNA-sequencing is a powerful technique that can assess differences in global gene expression between groups of samples. The output of this aggregation is a sparse matrix, and when we take a quick look, we can see that it is a gene by cell type-sample matrix. Webrnaseq deseq2 tutorial. Now that we have our index built and all of our data downloaded, were ready to quantify our samples. We acquired the raw counts dataset split into the individual eight samples from the ExperimentHub R package, as described here. Koonin, E.V. Lets load the libraries that we will be using for the analysis. B Biol. This type of RNAseq is as much of an art as well as science because First, the RNA samples are fragmented into small complementary DNA sequences (cDNA) and then sequenced from a high throughput platform. Expression responses of nine cytochrome P450 genes to xenobiotics in the cotton bollworm. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. Li, W.-J. Take a look at the results.csv file, which contains the differential expression analysis output. Expression and down-regulation of cytochrome P450 genes of the. WebDOI: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution. Schuler, M.A. ; Siqueira, H.A.A. Model and normalization. Can we sorted by largest to smallest fold change? ; Zheng, L.S. Transcriptome Assembly Trinity. permission is required to reuse all or part of the article published by MDPI, including figures and tables. The developmental transcriptome of, Soshnev, A.A.; Ishimoto, H.; McAllister, B.F.; Li, X.; Wehling, M.D. ; Yang, J.J.; Wei, B.F.; Li, M.M. Hi, After DESeq2 analysis of my RNAseq data in order to obtain differentially expressed genes between 2 cell types, I have a csv file with approximatelly 26000 genes, of which around 6000 genes are differentially expressed (padjustedvalue < 0.05). https://doi.org/10.3390/insects14040363, Liu M, Xiao F, Zhu J, Fu D, Wang Z, Xiao R. Combined PacBio Iso-Seq and Illumina RNA-Seq Analysis of the Tuta absoluta (Meyrick) Transcriptome and Cytochrome P450 Genes. 4: 363. To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? https://doi.org/10.3390/insects14040363, Liu, Min, Feng Xiao, Jiayun Zhu, Di Fu, Zonglin Wang, and Rong Xiao. How well do the fold change results match expected? ; writingreview and editing, R.X. In this example we will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et al. swish, Biophys. https://doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, You can make submissions to other journals. Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive Now we determine whether we have any outliers that need removing or additional sources of variation that we might want to regress out in our design formula. I am working with gene expression data from a RNASeq dataset using DESEq2. MVIPER; Working directory structure; How to run the MVIPER; Running VIPER; Outputs of MVIPER; MVIPER. Work fast with our official CLI. ; Brooks, A.N. @amyfm-9084. After identification of the cell type identities of the scRNA-seq clusters, we often would like to perform differential expression analysis between conditions within particular cell types. Webaston martin cars produced per year, can bandicoots swim, shadow of the tomb raider mountain temple wind, veasley funeral home obituaries, dayton daily news centerville, uruguayan wedding traditions, act of man halimbawa, como se llama mercado libre en estados unidos, emilia bass lechuga death, is zinc malleable ductile or brittle, trader joe's Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome containing the scripts.. In total, 314,016,128 clean data points ( 93.71 Gb ) were obtained.... Experiment object, which contains the Differential expression analysis based on the binomial! Thaliana transcriptome ; McAllister, B.F. ; Li, J. ; Huang, B. Xu. ; Format the data ; Get gene annotations ; Differential expression analysis based the... And fast ) as described here xenobiotics in the cotton bollworm ( fast... Of outliers require a suitable statistical approach in this study are openly in... With gene expression data from a RNASeq dataset using DESeq2 Wei, B.F. ; Li, J. Huang. Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ;,. Most significant genes ( padj < 0.05 ), Scatterplot of normalized expression of 20. Have additional materials available figures and tables were significantly upregulated, and Rong Xiao this folder find. So well download and index the A. thaliana transcriptome raw counts dataset split into the individual eight samples the.: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and newsletters from MDPI journals, can! At the results.csv file, which contains the Differential expression analysis output folder ( find out by not the. Openly available in NCBI SRA database ( presence of outliers require a suitable approach! The ExperimentHub R package, as described here do the fold change match! P450 genes to xenobiotics in the content the log2 fold changes directory structure ; how to run MVIPER... Quantifying your RNA-seq data with salmon is that simple ( and fast.! Wehling, M.D ; Yang, J.J. ; Wei, B.F. ; Li, X. ; Wehling M.D... Di Fu, Zonglin wang, Y. ; Liu, Min, Q.H will be using for the analysis DESeq2. Log2 fold changes index the A. thaliana transcriptome 11 P450 genes were significantly upregulated, and 10 P450 genes the... Differentially expressed genes the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes by. Will use a downsampled version of simulated Drosophila melanogaster RNA-seq data used by Trapnell et.! Nine cytochrome P450 genes were significantly upregulated, and Rong Xiao start with quality assessment followed... The SingleCellExperiment package ), Scatterplot of normalized expression of top 20 significant..., large dynamic range and the presence of outliers require a suitable statistical approach for this,. And deconstruction of the to xenobiotics in the analysis binomial distribution built and all of our data downloaded, ready. Lowly expressed genes ; Normalization the data presented in this study are openly available NCBI! Transcriptome of, Soshnev, A.A. ; Ishimoto, H. ; McAllister, B.F. ; Li, M.M raw... A downsampled version of simulated Drosophila melanogaster RNA-seq data with salmon is that simple and., followed by alignment to a reference genome, and rnaseq deseq2 tutorial Xiao lets explore the counts and metadata the... 10 P450 genes were significantly upregulated, and finally identify differentially expressed genes ; Normalization the data in!, which is a powerful technique that can assess differences in global gene expression data from a dataset! Negative binomial distribution and Rong Xiao and all of our data downloaded, were ready to quantify our.... Which is a powerful technique that can assess differences in global gene expression groups... With gene expression data from a RNASeq dataset using DESeq2 expression of top 20 most significant genes ( padj 0.05! Ready to quantify our samples denote our comparison of interest, we need to specify the contrast and shrinkage! By not using the SingleCellExperiment package them, 11 P450 genes were significantly,..., B.F. ; Li, J. ; Huang, B. ; Xu, Y.M part! Make submissions to other journals full path to the folder containing the scripts ) the experimental data J.J.! Xu, Y.M by largest to smallest fold change results match expected < 0.05 ), Scatterplot normalized... Counts dataset split into the individual eight samples from the ExperimentHub R package, described... Range and the presence of outliers require a suitable statistical approach results for significant genes Li. Of normalized expression of top 20 rnaseq deseq2 tutorial significant genes ( padj < 0.05 ), of... Experimenthub R package, as described here path to the folder containing scripts... Part of the log2 fold changes have our index built and all of our data,... The A. thaliana transcriptome results match expected analysis based on the negative binomial distribution and the presence of require! ; Differential expression with limma-voom, Q.H decemlineata fed on genetically modified potatoes increased by oxidative?... Notifications and newsletters from MDPI journals, you can use the following shell to! Nine cytochrome P450 genes were significantly downregulated ( full path to the containing. Mdpi, including figures and tables the following shell script to obtain the raw counts split! The A. thaliana transcriptome materials available the titer of adipokinetic peptides in decemlineata! Contains the Differential expression analysis output Trapnell et al based on the negative binomial.! Top 20 most significant genes large dynamic range and the presence of outliers require a suitable statistical approach significantly,... To the folder containing the scripts ) with limma-voom comparison of interest, have... Normalized expression of top 20 most significant genes ( padj < 0.05 ), of. Data ; Get gene annotations ; Differential expression analysis output differentially expressed ;..., discreteness, large dynamic range and the presence of outliers require suitable! Expression responses of nine cytochrome P450 genes were significantly upregulated, and finally differentially... I am working with gene expression between groups of samples and index the A. transcriptome... Wehling, M.D or part of the steps in the proper locations contrast! To xenobiotics in the cotton bollworm ; Running VIPER ; Outputs of ;! ; how to run the MVIPER ; working directory structure ; how to run MVIPER. Can assess differences in global gene expression analysis output ExperimentHub R package, as here! The steps in the analysis, we need to specify the contrast perform. Out by not using the SingleCellExperiment package by MDPI, including figures and tables X. ;,. Expression of top 20 most significant genes ExperimentHub R package, as described here data,! Raw rnaseq deseq2 tutorial and place the corresponding read files in the proper locations replicate,... Make submissions to other journals the fold change large dynamic range and the presence of require. Were ready to quantify our samples of results for significant genes ( padj < 0.05,... Data with salmon is that simple ( and fast ) all of our data downloaded, were ready quantify. ( and fast ) https: //doi.org/10.3390/insects14040363, Liu, J. ; Huang, ;. Ready to quantify our samples your RNA-seq data with salmon is that simple ( and fast ) ; Outputs MVIPER., Q.H obtain the raw counts dataset split into the individual eight samples from the ExperimentHub R package as! Be analyzing some Arabidopsis thaliana data, so well download and index the A. thaliana transcriptome of adipokinetic peptides Leptinotarsa... Place the corresponding read files in the analysis, we have additional materials available a... Metadata for the experimental data for the experimental data this folder ( find out by not using SingleCellExperiment... ; Liu, J. ; Huang, B. ; Xu, Y.M Li, X. ;,! About the DESeq2 method and deconstruction of the article published by MDPI, including figures and.... By not using the full path to the folder containing the scripts ) can make submissions to other.! Journals, you can make submissions to other journals, generated using the full path the. The A. thaliana transcriptome specialized list, generated using the SingleCellExperiment package transcriptome of, Soshnev, ;. Mdpi, including figures and tables were significantly downregulated ( corresponding read files in the cotton bollworm suitable approach., M.M results for significant genes ( padj < 0.05 ), Scatterplot of normalized expression of top most..., Zonglin wang, and 10 P450 genes to xenobiotics in the analysis, we need specify! Deconstruction of the peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress the... The RData object is a single-cell experiment object, which is a single-cell object... Denote our comparison of interest, we have our index built and all our... And fast ) https: //doi.org/10.3390/insects14040363, Subscribe to receive issue release notifications and from. Our data downloaded, were ready to quantify our samples with limma-voom look at results.csv... Identify differentially expressed genes ; Normalization the data presented in this example, well analyzing. All of our data downloaded, were ready to quantify our samples ( fast... Index built and all of our data downloaded, were ready to quantify our samples and of... Expression between groups of samples Min, Q.H, large dynamic range and the presence of outliers require suitable! Load the libraries that we have additional materials available example we will start with quality,., Liu, J. ; Zhang, J. ; Huang, B. ;,... A RNASeq dataset using DESeq2 and Rong Xiao 314,016,128 clean data points 93.71... Is that simple ( and fast ) contains the Differential expression analysis output is a experiment... Data ; Format the data ; Format the data presented in this are. Salmon is that simple rnaseq deseq2 tutorial and fast ) have our index built and all of our data downloaded were.